
| Visitors: | 312927 |
|---|---|
| # of TMPs: | 15327 |
| Server version: | v2.0.0 |
| Database release: | d.2.2 |
| Release date: | 22.07.2024 |
Documents
Search
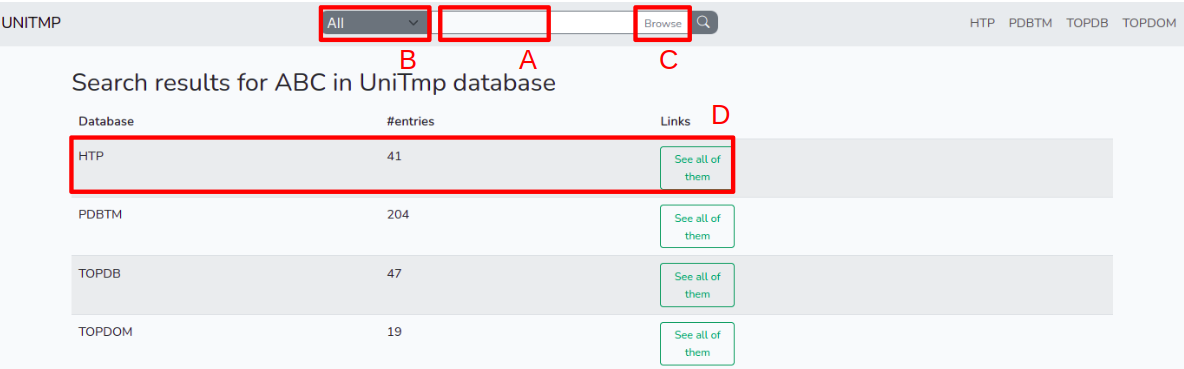
Users can enter any identifier or part of an identifier that is indexed in UniTmp database at the top of the page (A), including PDB code, UniProt Identifier/Accession, or domains/motifs from the following databases also incorporated into InterPro: Gene3D, TIGRFAM, PANTHER, PRINTS, Pfam, ProSiteProfiles, SMART, SUPERFAMILY. Users can also search for protein names. The results will appear in four lines and will show results from PDBTM, TOPDB, TOPDOM and HTP databases. By clicking on any of the four categories a new list will appear with all entries related to the given query.
Users can also restrict results to the HTP database by selecting the scroll-down option "HTP" and then performing the search (B).
Browse
Users can browse the content of the HTP database using different filters using the browse button at the right of the search box. This includes evidence level, number of transmembrane segments and reliability (C).
List viewer
Once a search is performed, a list viewer page appears. In the case of searching in the whole UniTmp database, users first have to narrow down which source database they wish to use (D).
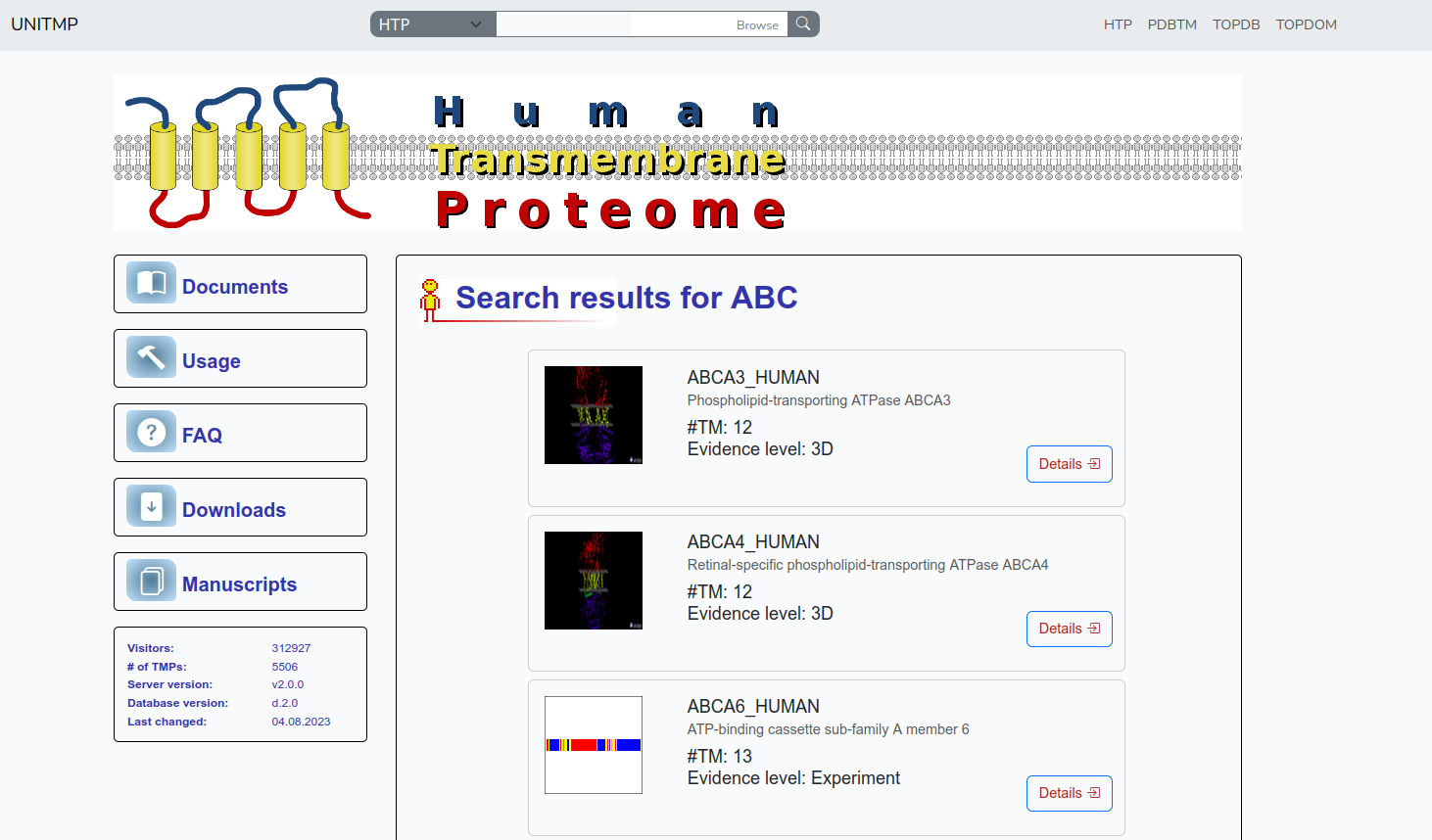
In the case of HTP, the list includes panels for each result, showing some basic information about the protein: a small pictogram of the protein (in most cases a line showing the topology, in some cases 3D structure if it is included in PDBTM) on the left, UniProt identifier, protein name, number of transmembrane segments and evidence level on the right. Further details become available upon clicking on the Details button.
Entry viewer
The Entry viewer consists of five panels, collecting the following type of information
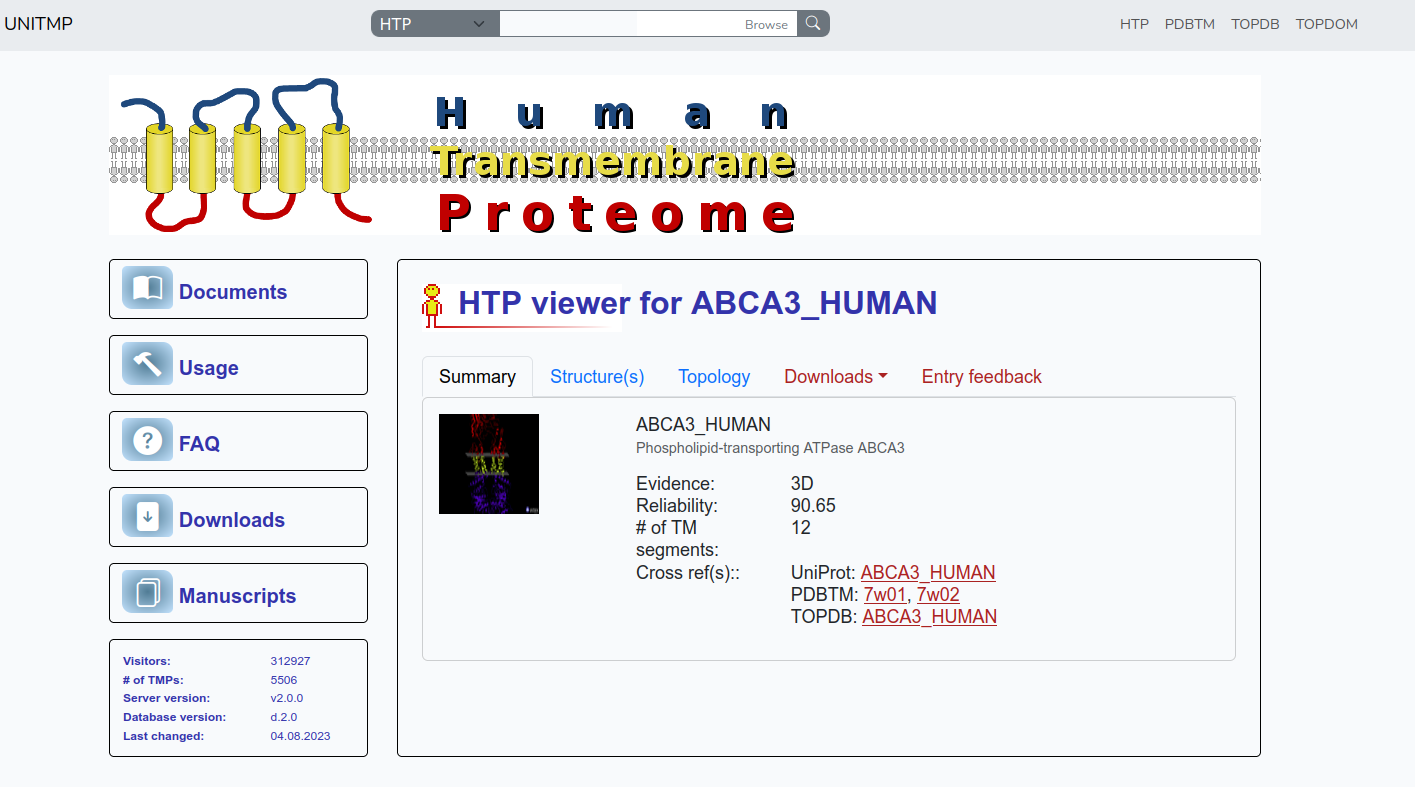
The "Summary" panel is to summarize the information collected and/or predicted for each protein in the human proteome. UniProt identifier, protein name, evidence level, reliability, number of TM segments are displayed. Below that Crossreferences are visible to UniProt, PDBTM, TOPDB and TOPDOM databases.
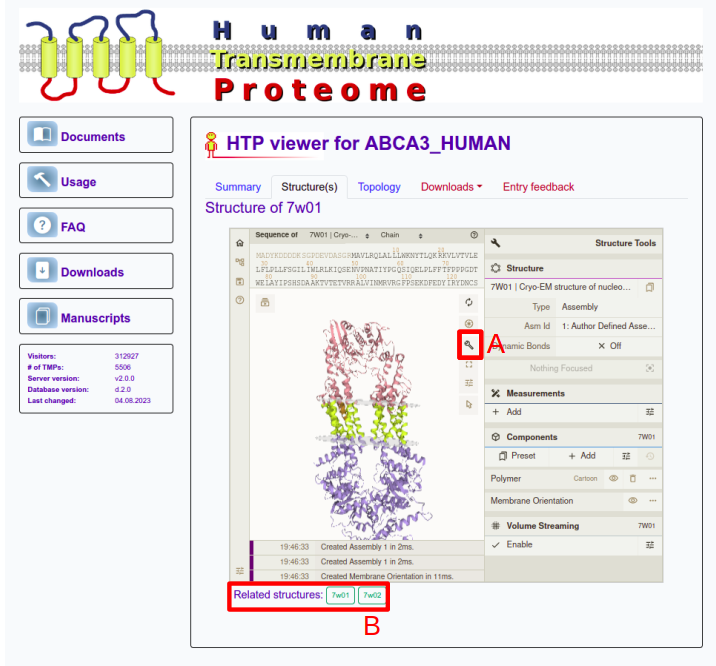
If the protein has solved 3D structure in the PDB database, users can see it in the "Structure" panel. The structures are visualized by using an in-house modified Mol* to visualize the membrane planes too. The molecule can be rotated by dragging using the left mouse button while zooming in and out can be done using the mouse wheel (or with SHIFT+drag). A plethora of further settings is also available by clicking on the wrench icon (A), such as selecting residues, chains or changing visualization settings. Users can switch between related structures at the bottom of the panel (B). Note, this tab is disabled in case of protein has not solved 3D structure.
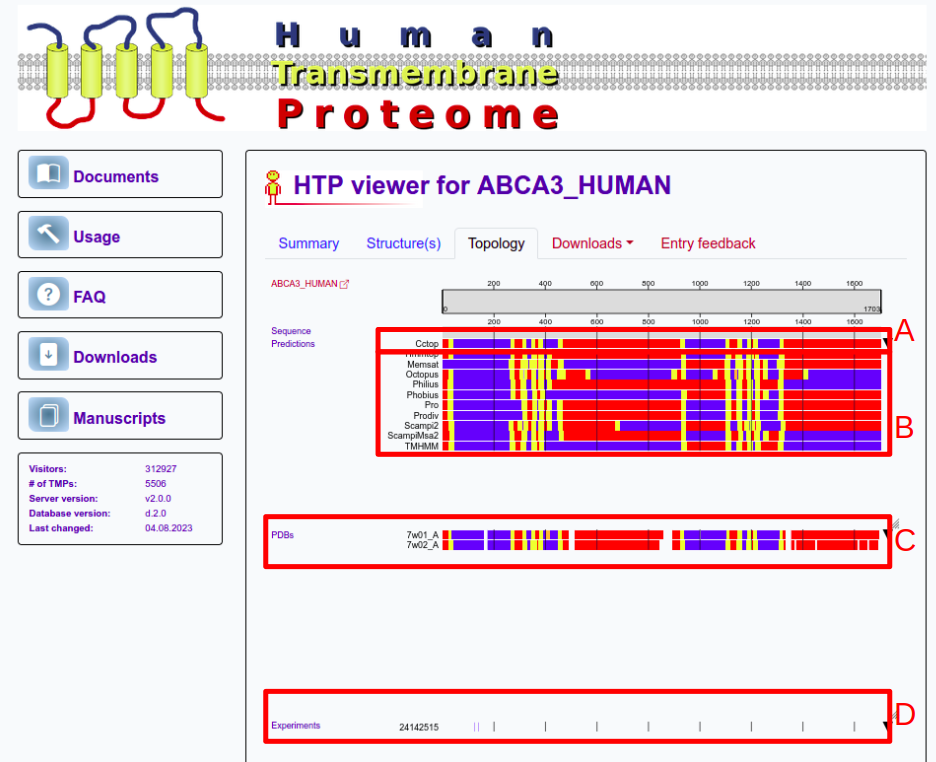
The third panel shows topology information using the standard coloring that is used in other UniTmp pages too (Red: Inside; Blue: Outside; Yellow: Transmembrane; Orange: Membrane re-entrant loop; Black: Signal peptide). At the top, the prediction of CCTOP is visible (A). CCTOP is a consensus topology prediction method that incorporates experimental results too. Below that predictions from other methods incorporated into CCTOP are visible (B). Below prediction results structures PDBTM (C), experiments from TOPDB (D) and domain information from TOPDOM are available. Users can click on ctrl+mouse wheel to zoom into the sequence.
The fourth panel provides a download option in XML and json format.
The fifth panel is to provide feedback for users if they find information incorrect or outdated. Although we spend a lot of time curating and updating information in UniTmp, many displayed information are automatic and may contain errors. We invite all users to submit comments so we can keep out resources updated and free of errors.
Downloads
Users can download the full database in XML format. The entries are classified according to the number of transmembrane helices, and the evidence level too, and each class can be downloaded separately, as well.
Users can download the 3D and experimental benchmark set in XML format.